
Click here to see all images
March, 2026
Case of the Month
Clinical History:A 30-year-old woman presented with a 7-day history of fever, cough, shortness of breath, and hemoptysis. She had short stature, microcephaly, dysmorphism with coarse facies , puffy eyes and thin telangiectatic skin. Seven months prior she was diagnosed as sarcoidosis based on her previous CT and high ACE levels and she was treated with prednisolone, mycophenolate, and azathioprine.
Current CT chest showed diffuse smooth lobular and interlobular septal thickening with GGOs in bilateral lungs (crazy paving pattern ) and multiple bilateral mediastinal lymphadenopathy CECT abdomen was done and it showed hepatosplenomegaly. Her lipid profile showed reduced HDL cholesterol (21 mg/dL), increased total cholesterol (194 mg/dL), and non-HDL cholesterol (176 mg/dL); CRP was increased. All other investigations including CBC, RFT, LFT, malarial parasite, blood cultures, and autoimmune profile were negative; her vitals and SpO2 were normal; 2D echo was normal.
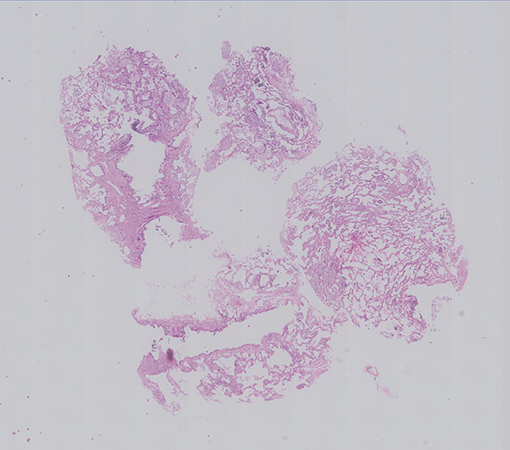
An EBUS guided Cryo lung biopsy and mediastinal lymph node biopsy were done along with Bronchial washings and sent for histopathological examination. Her BAL Infectious workup (bacterial, fungal, mycobacterial ) was negative. The cryobiopsy showed multiple fragments of tissue showing alveolated lung parenchyma with preserved architecture (Fig 1). Many clusters of foamy macrophages are noted in the alveolar spaces; they are variable in size, show multiple fine vacuoles in the cytoplasm with some of them showing deep blue granules (Fig 2). They are seen in the interstitium (Fig 3) as well as in the bronchovascular bundles. The alveolar interstitium is widened by moderate mononuclear inflammatory infiltrate and mild interstitial fibrosis.(Fig 2, 3). The bronchiolar epithelial cells are vacuolated, (Fig 3), however the Type II pneumocytes appear normal or show focal mild reactive atypia (Fig 2). No other significant pathological changes are identified.The pulmonary vasculature was unremarkable.
The mediastinal lymph node biopsy (Fig 4,5) and the bronchial wash cell block (Fig 6) also showed similar clusters of foamy macrophages A PAS and a PAS-D stains were done and the foamy macrophages were negative (Fig 7).
Based on the above histological features a diagnosis of Endogenous lipoid pneumonia was offered raising the possibility of Acid sphingomyelinase deficiency disease (ASMD). Enzyme activity assays and Genetic mutational studies were suggested for confirmation of the same.
Q1. 1. Which of the following Lysosomal storage disorder does not cause ILD?
- Fabry's disease
- Mucopolysaccharidosis
- Gaucher disease
- Acid sphingomyelinase deficiency
Q2. ASMD is caused by mutations in
- α-galactosidase gene (GLA)
- Glucocerebrosidase gene
- SLC34A2 gene
- Sphingomyelin phosphodiesterase 1 (SMPD1) gene
Q3. Intra alveolar foamy macrophages occur in all conditions except
- Hypersensitivity pneumonitis
- Gaucher's disease
- Vaping induced lung injury
- None of the above
Answers to Quiz
Q1. B
Q2. D
Q3. D
Q2. D
Q3. D
Diagnosis
Endogenous lipoid pneumonia: acid sphingomyelinase deficiency (ASMD, Niemann-Pick disease type B).
Discussion
Acid sphingomyelinase deficiency (ASMD), previously called Niemann–Pick disease is a rare autosomal recessive lysosomal storage disease caused by mutations in the sphingomyelin phosphodiesterase 1 (SMPD1) gene. It has three subtypes. Types A and B result from a deficiency of acid sphingomyelinase activity that lead to the accumulation of sphingomyelin in tissues of the reticuloendothelial system Type A ASMD presents as a fatal neurodegenerative disorder of infancy. Type B ASMD is a less severe form with milder neurological involvement, characterized by hepatosplenomegaly, interstitial lung disease and hyperlipidemia Type C ASMD is a genetically distinct disorder; it is a complex lipid storage disorder caused due to defective intracellular trafficking of cholesterol resulting in secondary glycosphingolipid accumulation; the clinical presentation is dominated by neurological involvement. Pulmonary involvement occurs in all three types of ASMDs, but most frequently in Type B.
The most common HRCT findings include ground-glass opacities , interlobular septal thickening and intralobular lines as well as superimposed ground glass on interlobular septal thickening (crazy paving pattern).
The lung biopsy shows diffuse infiltration of the alveoli, alveolar interstitium , lymphatics and subpleural spaces with lipid laden macrophages called Niemann–Pick cells or sea-blue histiocytes characterizing a type of endogenous lipoid pneumonia . These cells are large multivacuolated histiocytes containing fine and coarse granules that stain deep blue with May-Grunwald–Giemsa stain. There is vacuolation of bronchial ciliated epithelium without vacuolation of Type 2 pneumocytes. There may be mild interstitial inflammation and fibrosis but the underlying architecture is preserved . BAL specimens also demonstrate these characteristic cells.
Extra pulmonary manifestations include hepatosplenomegaly , thrombocytopenia and anemia, spleen infarction, cirrhosis, primary central nervous system disease , retinal abnormalities and bone disease in the form of osteopenia or osteoporosis. The Differential diagnosis includes the other Lysosomal storage disorders particularly Gaucher’s disease. Diagnosis of ASD is established by detection of: 1) low residual enzyme activity (ASM) in leukocytes; and 2) SMPD1 gene mutation analysis showing biallelic pathogenic variants.
Treatment : For Type A ASMD , the treatment is mainly symptomatic while Olipudase alfa, a recombinant human acid sphingomyelinase is available for Type B and Miglustat is available for Type C ASMD.
Take home message for trainees:
Lysosomal storage disorders should be kept in mind in the differential diagnosis of endogenous lipoid pneumonia, especially when the patient has systemic manifestations such as hepatosplenomegaly and hypercholesterolemia. Early diagnosis of these diseases is crucial to avoid irreversible organ damage and early initiation of Enzyme Replacement Therapy.
The most common HRCT findings include ground-glass opacities , interlobular septal thickening and intralobular lines as well as superimposed ground glass on interlobular septal thickening (crazy paving pattern).
The lung biopsy shows diffuse infiltration of the alveoli, alveolar interstitium , lymphatics and subpleural spaces with lipid laden macrophages called Niemann–Pick cells or sea-blue histiocytes characterizing a type of endogenous lipoid pneumonia . These cells are large multivacuolated histiocytes containing fine and coarse granules that stain deep blue with May-Grunwald–Giemsa stain. There is vacuolation of bronchial ciliated epithelium without vacuolation of Type 2 pneumocytes. There may be mild interstitial inflammation and fibrosis but the underlying architecture is preserved . BAL specimens also demonstrate these characteristic cells.
Extra pulmonary manifestations include hepatosplenomegaly , thrombocytopenia and anemia, spleen infarction, cirrhosis, primary central nervous system disease , retinal abnormalities and bone disease in the form of osteopenia or osteoporosis. The Differential diagnosis includes the other Lysosomal storage disorders particularly Gaucher’s disease. Diagnosis of ASD is established by detection of: 1) low residual enzyme activity (ASM) in leukocytes; and 2) SMPD1 gene mutation analysis showing biallelic pathogenic variants.
Treatment : For Type A ASMD , the treatment is mainly symptomatic while Olipudase alfa, a recombinant human acid sphingomyelinase is available for Type B and Miglustat is available for Type C ASMD.
Take home message for trainees:
Lysosomal storage disorders should be kept in mind in the differential diagnosis of endogenous lipoid pneumonia, especially when the patient has systemic manifestations such as hepatosplenomegaly and hypercholesterolemia. Early diagnosis of these diseases is crucial to avoid irreversible organ damage and early initiation of Enzyme Replacement Therapy.
References
von Ranke FM, Pereira Freitas HM et al.Pulmonary Involvement in Niemann-Pick Disease: A State-of-the-Art Review. Lung. 2016 Aug;194(4):511-8. doi: 10.1007/s00408-016-9893-0. Epub 2016 May 10. PMID: 27164983.
Borie R, Crestani B, Guyard A, et al. Interstitial lung disease in lysosomal storage disorders. Eur Respir Rev 2021; 30: 200363 [DOI: 10.1183/16000617.0363-2020] .
Borie R, Crestani B, Guyard A, et al. Interstitial lung disease in lysosomal storage disorders. Eur Respir Rev 2021; 30: 200363 [DOI: 10.1183/16000617.0363-2020] .
Contributors
Dr. Sharada Nagoti MD
Consultant Thoracic and Lung Transplant Pathologist
C Path Labs, Mumbai, India
Dr Vijay Kumar Chennamchetty
Lead Interventional Pulmonologist
Apollo Health City , Hyderabad, India
Consultant Thoracic and Lung Transplant Pathologist
C Path Labs, Mumbai, India
Dr Vijay Kumar Chennamchetty
Lead Interventional Pulmonologist
Apollo Health City , Hyderabad, India