
Click here to see all images
April, 2024
Case of the Month
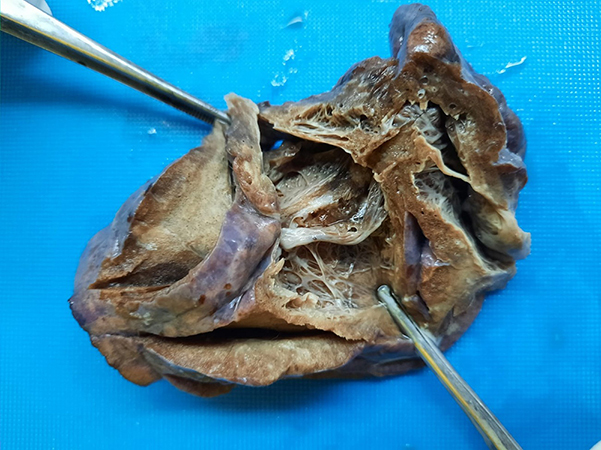
Clinical History: A 1 year old female presented with increasing respiratory distress and hemoptysis. The baby was born full term in an uncomplicated vaginal delivery. A CT scan demonstrated a 6.8 x 5.5 x 4.4 cm cystic lesion in right lower lobe. A right lower lobe lobectomy was completed, and grossly multiple cysts, measuring up to 7x5x5 cm (Fig 1) were identified. No nodules, masses or pleural thickening are seen. Histologic sections show multiple irregularly shaped cysts with branching papillae and lined by pseudostratified columnar epithelium. (Fig 2). The wall of the cyst shows bundles of smooth muscle fibers scattered mononuclear cells (lymphocytes and few plasma cells) and few lymphoid aggregates. (Fig 3, 4) The adjacent lung parenchyma shows focal areas of collapse (Fig 6) and emphysematous changes. (Fig 5). No mucinous cell proliferations or clusters are identified. There is no evidence of blastemal component or dysplasia or malignancy. No abnormal systemic arterial supply was identified.
Q1. What type of congenital pulmonary airway malformations (CPAM) most commonly have mucinous cell proliferation?
- Type 1
- Type 2
- Type 3
- Type 4
Q2. Which of the following CPAM types have been associated with mosaic activating KRAS mutations and an increased risk of metastatic mucinous adenocarcinoma?
- Types 0 and 4
- Types 1 and 2
- Types 1 and 3
- Types 2 and 3
Q3. What type of CPAM are now favored to represent cystic pleuropulmonary blastomas?
- Type 1
- Type 2
- Type 3
- Type 4.
Answers to Quiz
Q2. C
Q3. D
Diagnosis
Discussion
CPAM were earlier termed as Congenital Cystic Adenomatoid Malformation (CCAM) but later the terminology was changed to CPAM as not all these malformations are cystic or adenomatoid. In the initial classification scheme by Stocker, they described five major types on the basis of the area of malformation in the tracheobronchial tree. More recent classification systems typically recognize Type 0 and Type 4 CPAMs to represent other lung abnormalities. Acinar dysplasia is the current preferred term for the diffuse malformation that has been previously described as type 0 CPAM which is incompatible with life. Type 4 CPAMs are now classified as cystic pleuropulmonary blastoma and can often be associated with germline mutations in DICER1.
Recent studies to understand the basis of the remaining types of CPAM have identified mosaic activating mutations in KRAS in Type 1 and type 3 CPAMs. In these types there are increased rates of mucinous adenocarcinoma arising from the mucinous cell proliferations that can be seen in up to 75% of type 1 CPAMs and in up to 45% of type 3 CPAMs. Type 2 CPAMs are thought to arise secondary to bronchial atresia and have not been associated with KRAS mutations.
CPAM Type 1 is the most common type of CPAM and typically has large single or multiple cysts that can be recognized grossly. These cysts can range in size from <0.5 cm to greater than 10 cm. The cysts are lined by ciliated, pseudostratified columnar epithelium; segments of mucus-producing cells can be seen among the lining epithelial cells in most cases. The epithelium typically has an increased epithelial complexity with papillary projects and irregularly shaped small cyst spaces.
CPAM Type 2: is the 2nd most common type and is typically characterized by smaller cysts that can have a variable gross appearance of up to 2.5 cm. The cysts are lined by ciliated columnar epithelium and epithelial complexity is typically not observed. Mucinous proliferation is typically not seen.
CPAM Type 3: typically appear as solid or dense lesion on gross examination that is distinct from the uninvolved lung parenchyma. Histologically the cells resemble pulmonary hyperplasia or immature lung in the early canalicular stage of development with cuboidal to columnar epithelium. The septa often have prominent mesenchyme that along with the cuboidal epithelium results in a thickened appearance. Mucinous cell clusters can be seen in a subset of these lesions.
There are rare, reported cases of development of mucinous adenocarcinomas in patients with Type 1 or Type 3 CPAMs. These cases are typically seen in incomplete resection in infancy or resection in adulthood. In the histopathology report, the presence of mucinous epithelium and the completeness of resection should be documented for follow-up purposes.
The main differential diagnosis includes an intrapulmonary bronchogenic cyst which typically has a single thick-walled cystic lesion that does not connect to the adjacent alveolar spaces and typically has cartilage and submucosal glands in the wall. Pulmonary sequestration is also in the differential diagnosis, and it can have CPAM type 2 like changes. The key distinction is the presence of an abnormal systemic feeding vessel entering away from the hilum. Congenital lobar emphysema can present with respiratory distress and can be considered in the differential, however, shows uniform over distension of apparently normally developed acini with alveolar saccules and alveoli but are otherwise unremarkable, no cystic or solid components
The treatment for CPAMs is typically complete surgical resection and the long term prognosis for patients with complete resection in infancy or early childhood is very good.References
Husain, A.N., Stocker, T., Dehner, L.P. Stocker and Dehner’s Pediatric Pathology Fourth edition, 2021
Rosado-de-Christenson ML, Stocker JT. Congenital cystic adenomatoid malformation. Radiographics 1991;11:865–866. doi:10.1148/radiographics.11.5.1947321.
Stocker JT, Madewell JE, Drake RM. Congenital cystic adenomatoid malformation of the lung. Classification and morphologic spectrum. Hum Pathol. 1977 Mar;8(2):155-71. doi: 10.1016/s0046-8177(77)80078-6.
Giubergia V, Barrenechea M, Siminovich M, Pena HG, Murtagh P. Congenital cystic adenomatoid malformation: clinical features, pathological concepts and management in 172 cases. J Pediatr (Rio J). 2012;88(2):143–148. doi:10.2223/JPED.2177.
Nelson ND, Xu F, Chandrasekaran P, Litzky LA, Peranteau WH, Frank DB, Li M, Pogoriler J. Defining the spatial landscape of KRAS mutated congenital pulmonary airway malformations: a distinct entity with a spectrum of histopathologic features. Mod Pathol. 2022 Dec;35(12):1870-1881. doi: 10.1038/s41379-022-01129-0.
Langston C. New concepts in the pathology of congenital lung malformations. Semin Pediatr Surg. 2003 Feb;12(1):17-37. doi: 10.1053/spsu.2003.00001.
Dehner LP, Messinger YH, Schultz KA, Williams GM, Wikenheiser-Brokamp K, Hill DA. Pleuropulmonary Blastoma: Evolution of an Entity as an Entry into a Familial Tumor Predisposition Syndrome. Pediatr Dev Pathol. 2015 Nov-Dec;18(6):504-11. doi: 10.2350/15-10-1732-OA.1.
Contributors
Consultant Thoracic Pathologist
C Path Labs,
Hyderabad, India